Table Of Content
The space inside the ligand binding region would be studied with virtual probe atoms of the four types above so the chemical environment of all spots in the ligand binding region can be known. Hence we are clear what kind of chemical fragments can be put into their corresponding spots in the ligand binding region of the receptor. Thus, the induction principle should be respected to avoid overfitted hypotheses and deriving overfitted and useless interpretations on structural/molecular data.The SAR paradox refers to the fact that it is not the case that all similar molecules have similar activities. Bioisosteresare substituents or groups that have chemical or physical similarities, and which produce broadly similar biological properties. Bioisosterism is an important lead modification approach that has been shown to be useful to attenuate toxicity or to modify the activity of a lead, and may have a significant role in the alteration of pharmakinetics of a lead. Once a suitable target has been identified, the target is normally cloned and produced and purified.
Molecular Pharmacology and Toxicology (MPTX)
Why Is TikTok Parent ByteDance Moving Into Biology, Chemistry And Drug Discovery? - Forbes
Why Is TikTok Parent ByteDance Moving Into Biology, Chemistry And Drug Discovery?.
Posted: Wed, 03 Jan 2024 08:00:00 GMT [source]
Until now, this has often involved laborious manual work, and in many cases the search yielded molecules that were very difficult or impossible to synthesise. If researchers used AI in this process at all in recent years, it was primarily to improve existing molecules. Moving forward, we aim not only to encourage researchers studying computational methods related to drug screening to develop their own online platforms based on their research findings, but also to address the shortcomings of Drug-Online. Our objective is to create a fully functional and high-performance integrated online platform, which truly offers user-friendly services for biologists and pharmacists in the field of computational-based drug screening.
George Washington University
The rule was formulated by Christopher A. Lipinski in 1997, based on the observation that most medication drugs are relatively small and lipophilic molecules. Now, without human intervention, a generative AI is able to develop drug molecules from scratch that match a protein structure. This groundbreaking new process ensures right from the start that the molecules can be chemically synthesised.
Articles:
To characterize the various enzyme conformations involved in the isomerization of 1-phospho to 6-phosphohexoses, 15 high-resolution crystal structures of the phosphoglucomutase enzyme while performing the isomerization of glucose 1-phosphate to glucose 6-phosphate were obtained. Glucose 1,6-bisphosphate undergoes a 180° reorientation between the two phosphoryl transfer steps of the reaction. The enzyme with the phosphoserine bound to a Mg2+ ion has the same conformation at the beginning of the catalytic process, when it is bound to the substrate glucose 1-phosphate, and at the end of it, when it is bound to the product glucose 6-phosphate. During the reorientation of the sugar, when the catalytic serine is in the dephosphorylated state and bound to the glucose 1,6-bisphosphate intermediate, the enzyme has a different structure. In the future, the structure of such intermediates of the enzymes may suggest new drug molecules eventually able to trap, in these intermediate conformations, even the enzymes that are currently not druggable. NMR [25] and EPR [26] measurements can also feed data to molecular in silico calculations to determine the evolution of a protein with time, although limited to the active site or oligonucleotide structures.
For example, when a promising docked molecule is designed and modeled with favorable interactions with the target protein, it is compared to the active structures. Likewise, when an interesting mimic of an active compound is considered, it is docked into the protein to see if the two approaches lead to convergent conclusions. A crystallographic structure reveals the essential structural features of the active site. Specific intermolecular interactions can be incorporated in the ligand to strengthen its binding to the active site of the target protein. They believe that their study provided a new vision of the anti-tumor activity of EVO and RUT via 3D multicellular spheroids and cellular uptake through the fluorescence of compounds and may be helpful for drug screening and cytotoxicity studies. The success of any QSAR model depends on accuracy of the input data, selection of appropriate descriptors and statistical tools, and most importantly validation of the developed model.
After the proof of concept, the sequence-to-drug concept appears to be a promising direction for rational drug design. The maze of the drug discovery process is still very complex and challenging, even when only considering the small-molecule approach and no other promising approaches, such as those involving monoclonal antibodies or polynucleotides. Precision medicine, from drug discovery to the bedside, is the main concern nowadays. New powerful tools are made available almost every day, but medicinal chemists are still looking in every direction, from natural products [82] to sophisticated modeling [83], in search of new drug candidates complying with the new targets emerging from precision medicine needs. To further progress in the medicinal chemistry field, we need, in addition to new targets, a more accurate description of their different conformations and possibly of the evolution of the target structure with time during the biological process. This, combined with the knowledge of the genetic variants of the targets, will lead to an increased number and precision of the “magic bullets” that are drugs, and allow the progress of precision medicine.
Target identification of PPIs
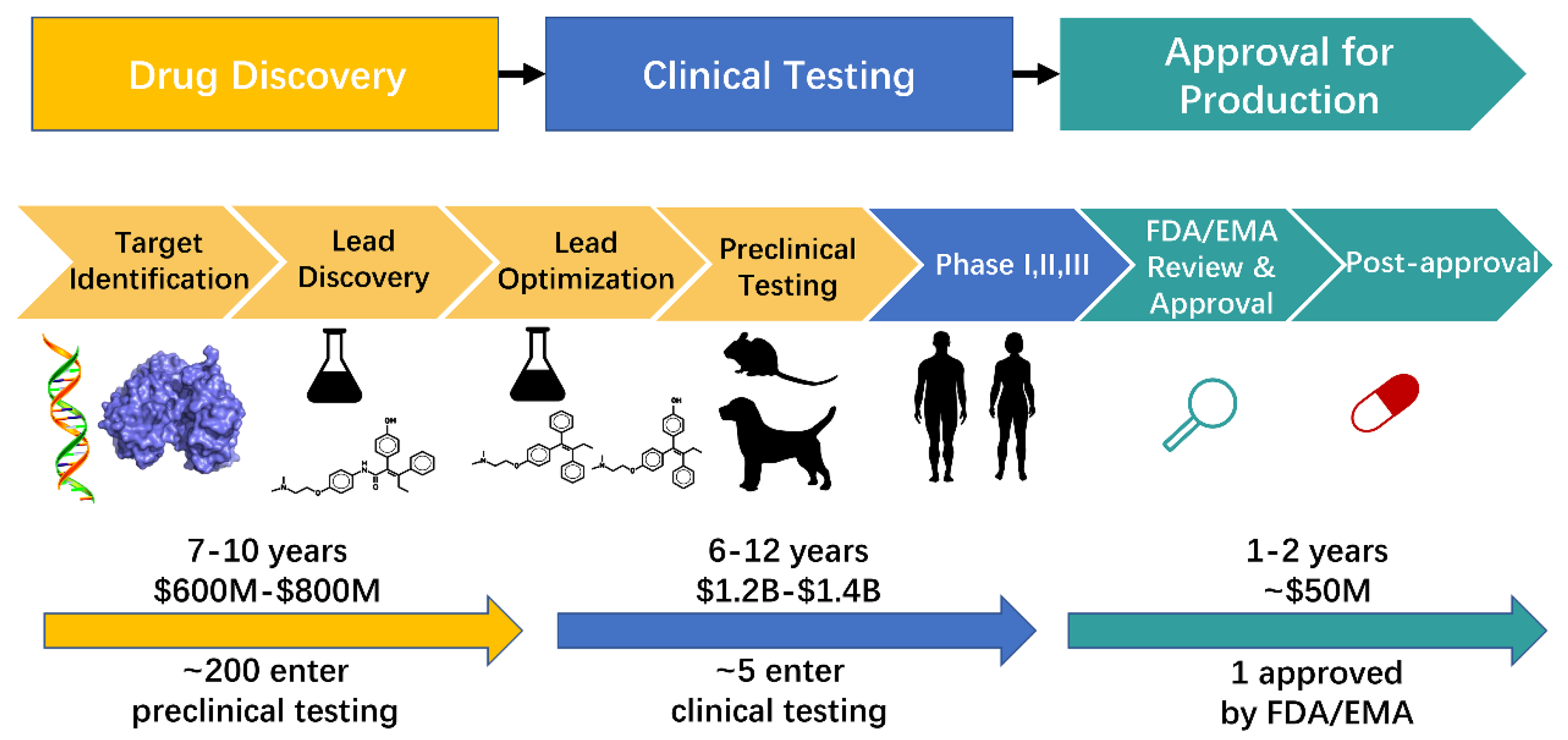
All new data discovered are collected on the Pharos website (pharos.nih.gov, accessed on 20 February 2022) [29]. From ten thousand synthesised and tested compounds, around 100 show some activity and safety, 10 of them enter the clinical trials and only 1 is approved for medical use [2]. Recent studies show that the initial number of tested compounds even passes one million [10]. However, as with the COVID-19 vaccines last year, we have seen that the time for drug development can be significantly reduced.

The program offers various specialization options, including Clinical Investigation, Evidence-based Medicine, and Health Services and Outcomes Research. In addition to a research based thesis, students are expected to complete 11 credits of core courses, six credits of research, and two and a half credits of electives. Though innovative projects are encouraged, the primary research strengths as of today (with the majority of faculty support) include oncology, neurological disorders/CNS diseases, and infectious diseases. Students enrolled full time are expected to complete this program in less than two years.
Fragmentary logP values have been determined statistically, based on empirical data for known logP values. This method gives mixed results and is generally not trusted to have accuracy of more than ±0.1 units. Straightaway the AI designed new molecules that also increase the activity of PPARs, like the drugs currently available, but without a lengthy discovery process.
ARF1 is a small G protein and belongs to the RAS superfamily, which switches between an active GTP-bound and an inactive GDP-bound conformation58. Inhibition of ARF1 activity is a promising direction for cancer immunotherapy, therefore, we selected ARF1 for investigation. The 2014 Ebola epidemic in West Africa is believed to have caused more than 11,000 fatalities. The request for novel drug development, finding efficient drug discovery pathways is going to be crucial in the fight against future outbreaks. They used the CANDO platform to generate top ranking drug candidates for Ebola virus disease treatment, which were compared to those identified from in vitro studies. They found that integrating computational docking predictions on a proteomic scale with results from in vitro screening studies may be used to select and prioritize compounds for further in vivo and clinical testing.
Rational drug design is only at the beginning of its development and is progressing rapidly. This indicates that new concepts and approaches will be soon in demand from computerized molecular design technologies. Rational drug design will be able to provide drug discovery with an entirely new dimension.
This challenging but necessary area engages students more intimately with the concepts of medical ethics, drug testing on human subjects, and the legal aspects of medicine. While the scientists’ ASO may well be an effective treatment, getting FDA approval for it would necessitate enormous clinical studies and could cost up to a billion dollars. The opinions expressed in all articles published here are those of the specific author(s), and do not necessarily reflect the views of Dove Medical Press Ltd or any of its employees. Drugdesign.org is a free access resource but in order to maintain it and develop further this project we need your helpso if you enjoy the content support us by donating to this project. Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and ... - Nature.com
Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and ....
Posted: Mon, 02 Oct 2023 07:00:00 GMT [source]
Sometimes bridging of the two carbon atoms (secondary cyclization) also leads to an increase in potency. Usually increasing the length of a saturated carbon side-chain from one (CH3) to 5 to 9 atoms (pentyl to nonyl) produces an increase in pharmacological effects.Further increase results in a decrease in the activity. This is probably either due to increase in lipohilicity beyond optimum value or decrease in concentration of free drug. Consequently, diazoxide( Hyperstat) was prepared as an antihypertensive drug without diuretic activity. A decrease in potency on removal of a group will suggest that it may have been pharmacophoric, an increase in potency means it was auxophoric and interfering with proper binding, and essentially no change in potency will mean that it is auxophoric but not interfering with binding. Once such pharmacophore is identified, structural modifications can be done to improve pharmacokinetic properties of the drug.








No comments:
Post a Comment